
Kraniopharyngeom im Kindes- und Jugendalter
Das Kraniopharyngeom – im Weiteren reden wir der Einfachheit halber vom Kranio - wird meist als Tumor bezeichnet, ist aber eigentlich keine Krebserkrankung. Wenn man Kraniogewebe untersucht, zeigen sich gutartige Gewebeeigenschaften, wie bei einer Fehlbildung. Diese Fehlbildung entsteht schon vor der Geburt. Im Laufe der Schwangerschaft wird aus einem kleinen Zellhaufen, der sich rasch teilt und entwickelt, nach 9 Monaten ein fertiges Baby. Wenn irgendwann während dieser Entwicklung Gewebe „aus dem Ruder läuft” und sozusagen nicht fertig wird, kann daraus später ein Kranio entstehen. Warum so was passiert, ist nicht bekannt. Man kann aber sicher sein, dass niemanden eine Schuld trifft.
Symptome und Behandlung
Der jüngste Patient, bei dem ein Kranio operiert wurde, war ein Neugeborenes. Bereits vor der Geburt sah man bei einer Ultraschalluntersuchung der Mutter die Erkrankung im Kopf des ungeborenen Kindes. Bei der Operation am 7. Lebenstag konnte dann ein großer Teil des Kranios entfernt werden (Abbildung 1).
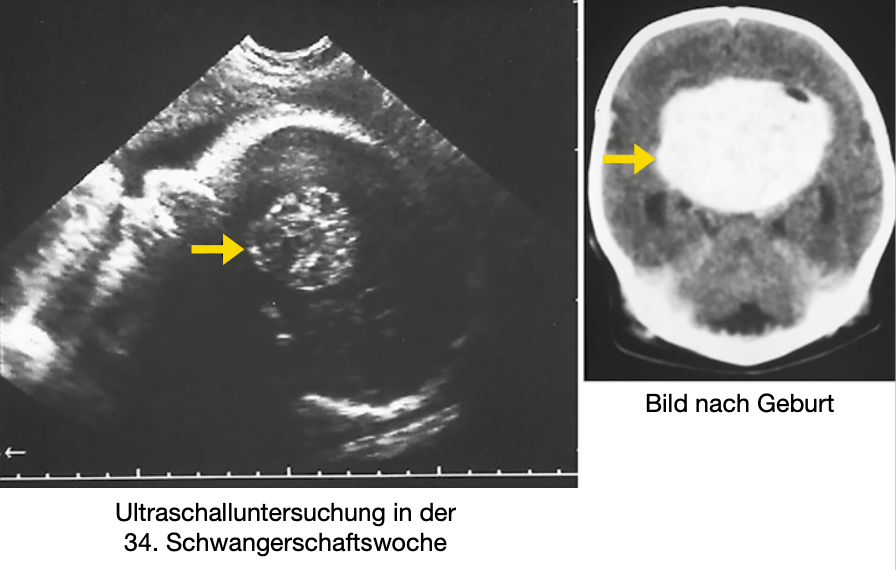
Obwohl das Kraniogewebe eigentlich gutartig ist, haben viele Betroffene Probleme, weil durch die Nachbarschaft zum Kranio andere Organe in Mitleidenschaft gezogen werden. Diese Probleme bestehen häufig schon lange, bevor die Diagnose gestellt wird. Viele Patienten klagen zum Teil über Jahre immer wieder über Kopfschmerzen. Hormonausfälle der Hirnanhangsdrüse führen dazu, dass oft das Wachstum eingeschränkt ist.
Bei einer Patientin fiel auf, dass sie viel zu früh – im Alter von 5 Jahren – in die Pubertät kam. Anfangs wusste keiner genau, warum bei ihr die Pubertätshormone verrückt spielten. Irgendwann wurde dann ein Kernspin (MRT) – eine Art Foto – von der Hirnanhangsdrüse gemacht und das Kranio entdeckt.
Nachdem die Diagnose im Kernspin gestellt ist, besteht die Behandlung meistens in einer Operation – schon allein, um sicher zu sein, dass es auch wirklich ein Kranio ist. Man hat lange Zeit geglaubt, dass alles wieder okay ist, wenn man es einfach ganz rausoperiert. Heute weiß man, dass trotz kompletter Entfernung Rückfälle möglich sind. Häufig verstecken sich nämlich einzelne Kraniozellen in der Umgebung, ohne dass man sie im Kernspin sieht. Und der Chirurg sieht diese Zellen bei der OP erst recht nicht.
Außerdem ist es ungesund, wenn bei der Operation andere Organe wie z. B. der Hypothalamus verletzt werden (Abbildung 2). Der Hypothalamus ist ein Teil des Gehirns (ganz in der Nähe zum Kranio), wo z. B. Appetit, Schlafrhythmus und viele andere Dinge reguliert werden, die man braucht, um gut durch den Alltag zu kommen. Der Hypothalamus steuert auch die Ausschüttung von Hormonen aus der Hirnanhangsdrüse, sodass es auch hier zu Problemen kommen kann. Eine Verletzung des Hypothalamus führt häufig zu einer raschen Gewichtszunahme nach der OP und einem Übergewicht, mit dem die Patienten auf Dauer zu kämpfen haben. Deswegen ist es bei der OP sinnvoll, keine komplette Entfernung zu planen, gerade wenn solche Verletzungen drohen. Ein schonendes Vorgehen ist erst recht wichtig, wenn schon vor der OP im Kernspin (MRT) zu sehen ist, dass das Kranio in den Hypothalamus vorgewachsen ist. Andererseits ist es leider oft so, dass die Hirnanhangsdrüse (Hypophyse) bei der Operation nicht geschont werden kann. Das heißt, die Drüse wird mit dem Kranio entfernt, weil beide zu sehr miteinander verwachsen sind. Aber die Hormone, die dort produziert werden, kann man gut durch regelmäßige Hormoneinnahme ersetzen – das ist eigentlich kein so großes Problem. Da wird man ganz automatisch im Laufe der Zeit sein eigener Spezialist, der sich mit den Medikamenten am besten auskennt.
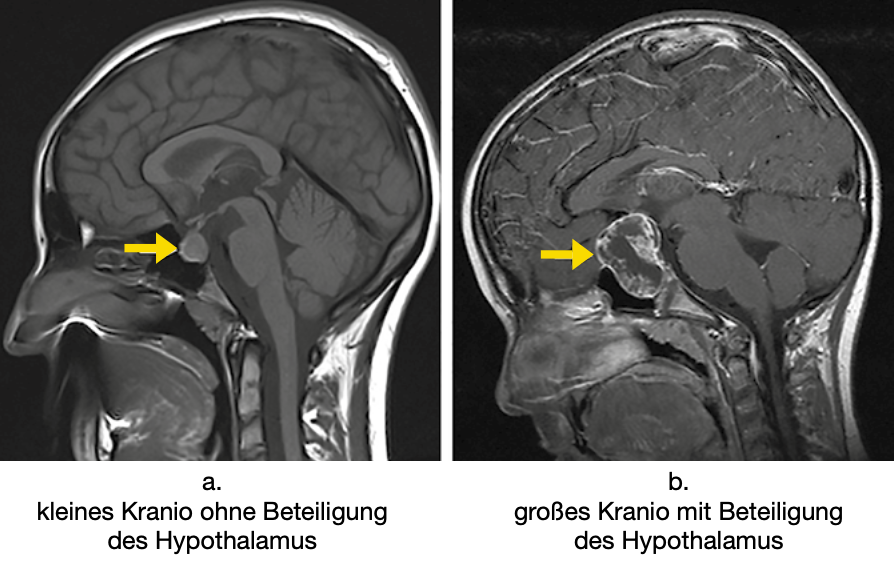
Was aber macht man, wenn es zu gefährlich ist, alles rauszuoperieren, mit dem Rest des Kranios, der nicht entfernt werden konnte? Nun, erst mal wartet man ab, ob der Kraniorest wieder größer wird. Also Kernspin (MRT), anfangs alle drei Monate. Wenn man dann leider wie so oft sieht, dass der Rest wieder wächst, müssen sich die Spezialisten beraten. Dann kommt eine Bestrahlung in Frage. Bestrahlt wird aber nur das verbliebene Kranio. Eine erneute OP ist oft deswegen erschwert, weil sich nach dem ersten Eingriff Narben gebildet haben, die es dem Chirurgen schwer machen, den Rest im zweiten Anlauf ganz zu entfernen.
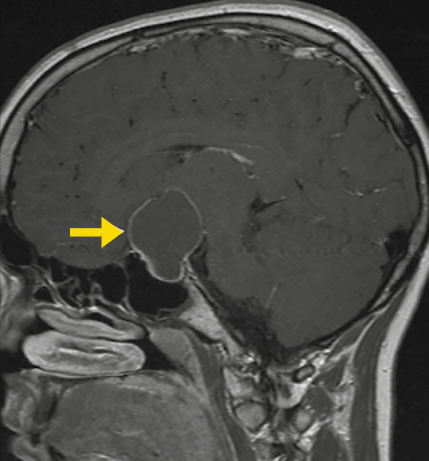
Manchmal besteht das Kranio aber auch aus einer großen Zyste (Abbildung 3). Eine Zyste muss man sich vorstellen wie einen mit Flüssigkeit gefüllten Ballon. Den Ballon muss man gar nicht rausoperieren – es reicht eigentlich, wenn man ihn anpiekst, dann fliesst die Flüssigkeit ab, die Zyste fällt in sich zusammen und macht dann keine Probleme mehr. Klingt gut – ist aber meistens nicht so. Das Loch, in das man reinpiekst, verschliesst sich oft ganz rasch wieder von selbst und die Zyste ist dann in alter Größe wieder da. Also muss man das Loch offenhalten, am besten mit einem Katheter, der mit einer kleinen Kapsel unter der Kopfhaut (Ommaya- Reservoir) verbunden ist. Über das Ommaya-Reservoir kann man dann, wenn nötig, immer wieder Flüssigkeit aus der Zyste rausholen und sogar Medikamente in sie spritzen (alpha Interferon). Sie verhindern, dass neue Zystenflüssigkeit gebildet wird.
Neue Perspektiven
Manchmal kommt es aber unweigerlich wegen der Lage des Kranios zu Spätfolgen, die für den Patienten sehr unangenehm sind. Störungen der Wachheit und des Gedächtnisses sowie eine ausgeprägte Gewichtszunahme beinträchtigen dann häufig das Wohlbefinden. Was kann man eigentlich dagegen tun? Die Behandlung ist deswegen so schwierig, weil es keine Medikamente gibt, die das Problem lösen. Bei ausgeprägter Müdigkeit und Störungen des Tag-Nacht-Rhythmuses kann es sinnvoll sein, eine Therapie mit zentralen Stimulantien (z. B. Methylphenidat) zu versuchen. Zur Behandlung der Adipositas, also der Fettleibigkeit, kommen Medikamente (GLP-1R-Agonisten) in Frage, die aber nur bei bestehendem Typ 2 Diabetes zur Behandlung zugelassen sind. Erste Untersuchungen haben gezeigt, dass Oxytocin günstige Effekte auf die Befindlichkeit und die Gewichtsentwicklung hat. Aber auch die Oxytocinbehandlung ist trotz ermutigender erster Ergebnisse noch nicht zugelassen. Als Betroffener braucht man also Geduld, was manchmal sehr schwer ist.
Auf einem wissenschaftlichen Gebiet gibt es in den letzten Jahren große Fortschritte. Man kann immer besser die Genetik, also die Erbinformation in Tumorzellen untersuchen. Die Untersuchungen wurden auch mit Kraniozellen gemacht. Bei einem bestimmten Kraniotyp konnte man zeigen, dass eine einzelne Störung vorliegt, die das Kranio zum Wachsen bringt. Ein Gen in der Erbinformation des Kranios - nämlich das BRAF-Gen - war an einer ganz bestimmten Stelle gestört. Kennt man das Gen und was es bewirkt, kann man heute mit einer gezielten Therapie (targeted therapy) den Effekt der Störung kurieren. Das ist auch bei einigen Patienten schon hervorragend gelungen. Das Wachstum konnte gestoppt werden. Das Kranio wurde kleiner und konnte dann besser operiert und bestrahlt werden. ABER diese Störung und Behandlungsmöglichkeit gibt es fast nur bei Betroffenen, die im Erwachsenenalter erkranken. Bei Kindern und Jugendlichen liegt fast immer ein anderer (adamantinomatöser) Typ vor, bei dem das BRAF-Gen gerade nicht gestört ist.
Prof. Dr. med. Hermann L. Müller,
Klinik für Allgemeine Kinderheil- kunde, Hämatologie/Onkologie,
Zentrum für Kinder- und Jugendmedizin,
Klinikum Oldenburg AöR,
Medizinischer Campus Universität Oldenburg